Up to now miniKar doesn't recognize chromosomes. It is used to build a training set for :
- supervised learning.
- features selection
miniKar can be configured with a configuration file which specify the path to the chromosomes images and the categories of of the training set (but also the size and positions of the red areas, representing the categories).
#read by miniChromClassif.py
[Fluorochromes]
Counter_stain=DAPI
Probes=Cy3,Cy5,FITC
[Images Path]
work_dir=/home/claire/Applications/ImagesTest/
user=jp
slide=Jpp48
field_list=13,14
[ClassifierGUI]
screen_size=1024,768
categories_number=4
categories_list=single chr,touching chr,nuclei,dusts
box_size=240,380
#((col,li),(col+200+30,li),...)!!
box_position=(20,300),(268,300),(516,300),(764,300)
From left to right, the red areas represents
- the single chromosomes.
- the overlapping chromosomes
- the nuclei
- dusts (or segmentation artifacts)
To use miniKar, three files must be in the same directory:
Suppose we want to classify the following particles into one of the four categories:
Suppose we want to classify the following particles into one of the four categories:
- single chromosome
- touching chromosomes
- nuclei
- dust
/home/user/cytogenetic_images/project/exampleslide/metaphase/DAPI/particles
Modify the configuration.cfg file to fit the path, for example:
work_dir=/home/user/cytogenetic_images
user=project
slide=exampleslide
field_list=1
user=project
slide=exampleslide
field_list=1
Run minikar.py from a console by : python minikar01.py or from your favorite IDE (spyder, ninja-ide,...). Something like this should be visible:

The green background is uggly, but the small things are visible. After having classified the different chromosomes, one can have :
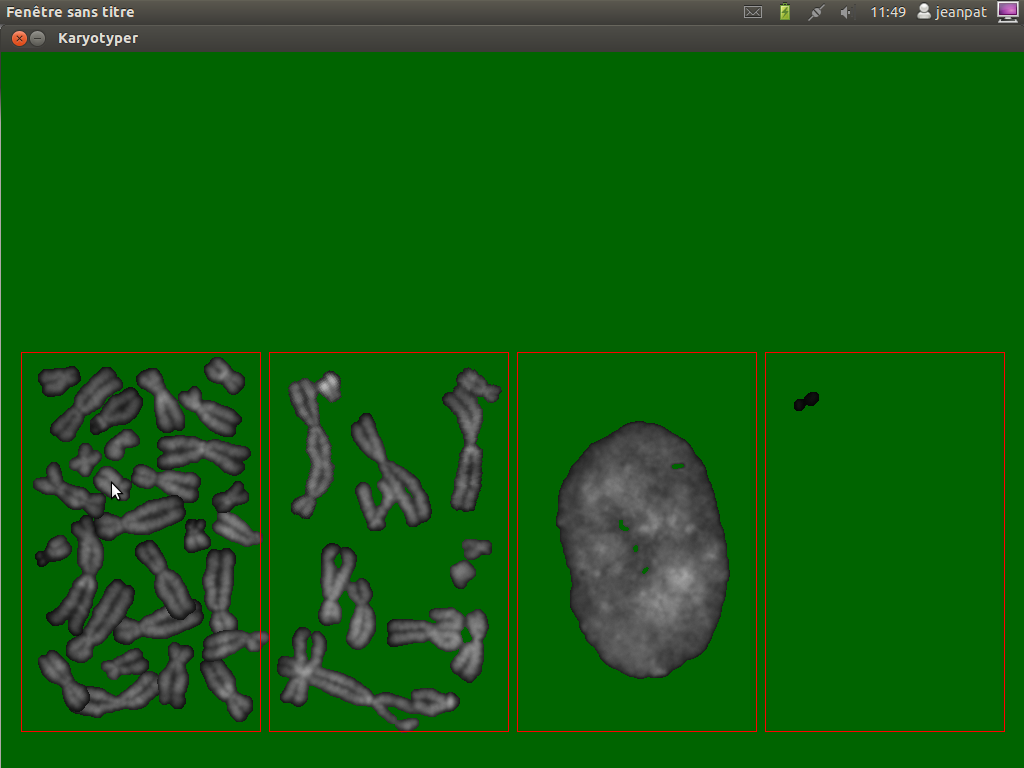
On pressing the keyboard Esc, minikar save a first file which associes the file name of a particle and its category and produce a second one in the csv format.
To classify the chromosomes from several metaphases, the configuration.cfg file must be modified for each metaphase:
To classify the chromosomes from several metaphases, the configuration.cfg file must be modified for each metaphase:
field_list=1
To classify the metaphase 13 in the folder exampleslide , modify the file as:
field_list=13
If features are computed for each particle, then they can be associated to a category for further processing with a classifier. Here, chromosomes from 14 metaphases were classified, five features related to the convex hull were used.
No comments:
Post a Comment